Epidemiologie
Die Erkrankung tritt weltweit auf und betrifft vorwiegend, aber nicht ausschliesslich, junge Erwachsene. Verlässliche epidemiologische Daten fehlen auf Grund der Seltenheit der Erkrankung. Berichtete Prävalenzen variieren zwischen 0,1-0,4 /100’000 Einwohner in Europa und Japan. Das mediane Alter bei Krankheitsbeginn liegt bei 36 Jahren 1.
Ätiologie/Immunpathologie
Die Ätiologie des Morbus Still des Erwachsenen (AOSD=Adult Onset Still Disease) ist unbekannt. Pathophysiologisch kommt es wahrscheinlich zunächst zu einer Aktivierung des innaten Immunsystems (neutrophile Granulozyten und Makrophagen). Dabei werden vermehrt Chemokine (CXCL 8), M-CSF und Interferon-g nachgewiesen. Aus Granulozyten freigesetzte S100 Proteine binden als ‘Alarmine’ an die sogenannten Pattern Recognition Rezeptoren und tragen zur Expression proinflammatorischer Zytokine wie Il-1, Il-6 und IL-18 via Inflammasom-Aktivierung bei 2.
Dieser Mechanismus erklärt die grosse Ähnlichkeit der Symptome zu den monogenetischen hereditären Fiebersyndromen sowie das gute Ansprechen der Erkrankung auf eine IL-1 oder IL-6 Blockade. Es wird postuliert, dass im Verlauf der Erkrankung das adaptive Immunsystem (vor allem T-Zellen) mit involviert wird. Möglicherweise ist das der Grund für die oft erst im Verlauf auftretende destruierende Arthritis. IL-18 und IL-1 können die Differenzierung zu proinflammatorischen Th-17 Zellen fördern 3. Es wird angenommen, dass diese Entwicklung durch eine frühzeitige Therapie (window of opportunity) beeinflusst werden kann. Des Weiteren wurden hohe Konzentrationen von IL-18 mit dem Auftreten eines Makrophagenaktivierungssyndroms (HLH) assoziiert, IL-18 fördert die Aktivierung von γδ-T Zellen und stimuliert natürliche Killerzellen (NK). Ein durch INF-γ induzierter ‘Zytokinsturm’ ist zentral in der Pathogenese des MAS.
Klinische Manifestationen
Der AOSD ist eine seltene Erkrankung, die typischerweise mit Fieber, Polyarthritis und Exanthemen einhergeht. Weitere häufige Symptome sind Halsschmerzen und Myalgien. Das Fieber folgt meist einem zirkadianen Rhythmus mit abendlichen Spitzen. Typisch ist der plötzliche Beginn.
In der Untersuchung findet sich gelegentlich eine Lymphadenopathie oder eine Hepatosplenomegalie. Eine Perikarditis tritt in einem Drittel der Patienten auf, seltener auch eine Pleuritis.
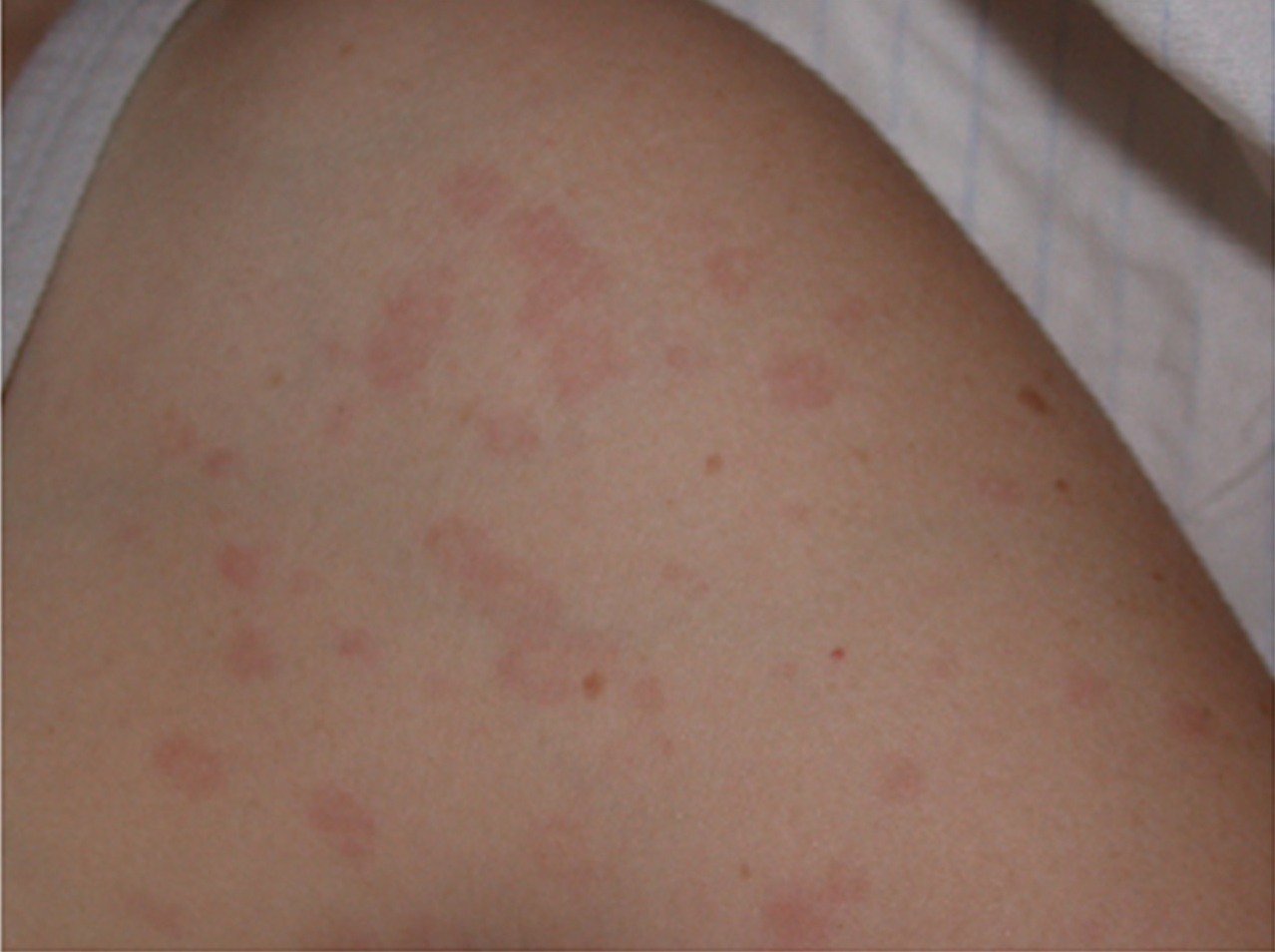
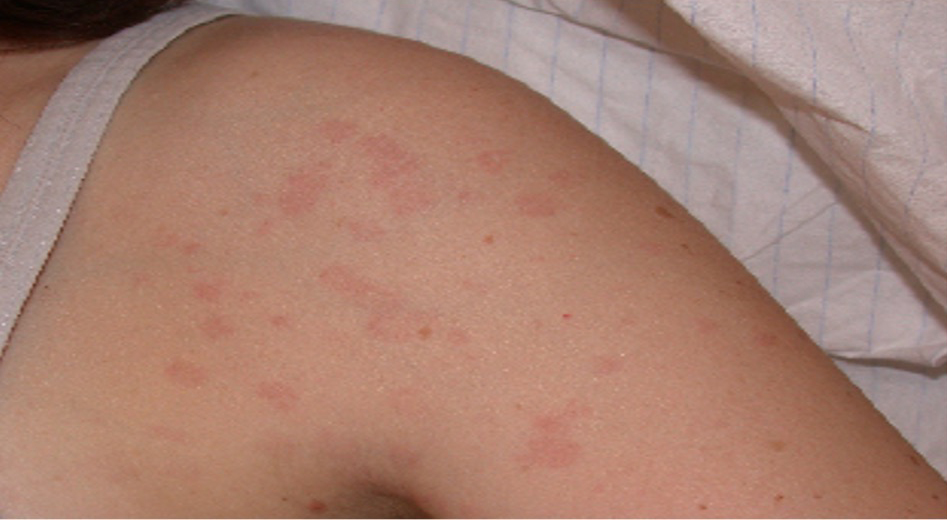
Das Exanthem ist oft flüchtig und kann mit dem Fieberanstieg assoziiert sein (Abb. 1). Aber auch urtikarielle Hautveränderungen können auftreten.
Die Arthritis ist oligo- oder polyartikulär; sie kann destruktiv verlaufen und unter anderem zu einer Destruktion der kleinen Handgelenke bis zum ‘os carpale’ (Foto) führen. Die gefürchtetste Komplikation ist das Makrophagenaktivierungs- Syndrom (HLH). Dieses tritt bei AOSD seltener auf als bei der systemischen juvenilen idiopathischen Arthritis. Selten wird auch eine pulmonal arterielle Hypertonie insbesondere bei AOSD Patienten mit hoch entzündlichem Verlauf beschrieben.
Charakteristische Laborwerte sind eine Leukozytose (Neutrophilie), eine Anämie und eine Thrombozytose im Rahmen der Entzündung sowie eine BSG- und CRP-Erhöhung. Typisch ist eine Hyperferritinämie (>5x des Normwertes). Des Weiteren finden sich gehäuft Transaminasenerhöhungen. Die Messung der glykolisierten Fraktion des Ferritins ist insbesondere bei sehr hohen Ferritinwerten diagnostisch hilfreich. Beim AOSD soll die Fraktion <20% des Gesamtferritins ausmachen.4
AOSD kann monophasisch, intermittierend oder chronisch verlaufen. Für die Diagnosestellung werden häufig die Yamaguchi Kriterien 2 verwendet (Tabelle 1). Wichtig ist aber der Ausschluss anderer Erkrankungen.
Tabelle 1: Yamaguchi Kriterien
Major Kriterien
Fieber >39°C >1 Woche
Arthralgien/Arthritis >2 Wochen
Typisches Exanthem
Leukozyten >10’000/ul,>80% Neutrophile
Minor Kriterien
Halsschmerzen
Lymphadenopathie
Erhöhung der Transaminasen
RF und ANA negativ
Mehr als 5 Kriterien, davon 2 Hauptkriterien, müssen für die Diagnose AOSD erfüllt sein.
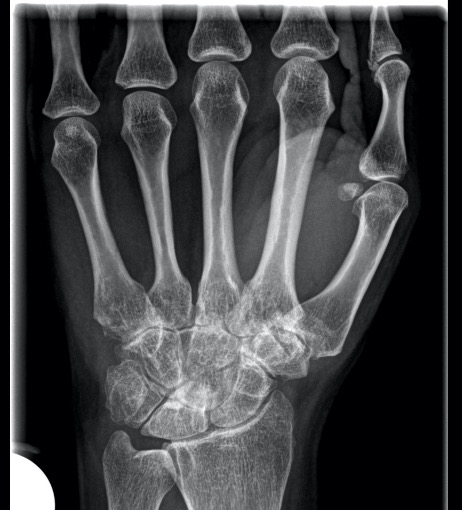
Es wird beobachtet, dass das AOSD im Verlauf den Charakter ändern kann. Auf die initiale hochinflammatorische Phase, gelegentlich ohne oder nur mit milder Arthritis, kann im Verlauf eine zweite Krankheitsphase folgen, bei der die arthritischen Beschwerden im Vordergrund stehen und die systemische Entzündungsreaktion nicht mehr so ausgeprägt ist 3. Radiologisch zeigen sich, vor allem im Verlauf, destruierende Gelenkveränderungen und Gelenkspaltverschmälerungen typischerweise an den karpometakarpalen Gelenken (Abb. 2). Die Autoantikörper sind meist negativ.
Die Differenzialdiagnose des AOSD ist breit. Es fehlen spezifische Symptome oder Marker. Die Diagnose sollte den Ausschluss anderer ähnlicher Erkrankungen umfassen. Die häufigsten Differenzialdiagnosen sind in Tabelle 2 aufgeführt.
Tabelle 2: AOSD Differenzialdiagnosen
| Erkrankung | Symptome | Untersuchung |
| Virale Infekte : HIV/EBV/SARS-COV-2, Parvo B-19 | Fieber, Lymphadenopathie, Hepatitis | Serologie, PCR |
| Sepsis, Endokarditis | Herzgeräusch | Kultur, Echo |
| Hämatologische Neoplasien | Lymphadenopathie, B-Symptome | PET/CT Differentialblutbild |
| DRESS Syndrom (Medikamenten Reaktion) | Eosinophilie, Fieber, Hepatitis | Medikamentenanamnese |
| Vaskulitiden/RA | Haut Niere Lunge | Auto AK |
| Hereditäre Fiebersyndrome | Periodizität | Genetik Familienanamnese |
Behandlung
Die Therapie sollte an den Schweregrad der Erkrankung angepasst werden. Selten ist bei sehr milden Verläufen eine Therapie mit NSAR oder tiefdosierten Steroiden möglich. Bei aktiverer Erkrankung, Organbeteiligung (z.B. Perikardtamponade) oder bei HLH werden initial hochdosierte Steroide eingesetzt. Aufgrund der zentralen Rolle der proinflammatorischen Zytokine der IL-1 Familie und IL-6 sind diese in der initialen autoinflammatorischen Phase das bevorzugte Ziel. Randomisierte Studien zur Therapie des AOSD sind rar auf Grund der Seltenheit der Erkrankung. Allerdings gibt es etwas mehr Daten aus Studien zur systemischen juvenilen idiopathischen Arthritis, die sowohl für eine IL-1 Blockade mit Canakinumab und Anakinra sowie für eine IL-6 Blockade mit Tocilizumab in randomisierten Studien eine Überlegenheit gegenüber Placebo gezeigt haben 8.
In der Schweiz ist der humane IL-1 b Antikörper Canakinumab seit 2014 für die Behandlung der systemischen juvenilen idiopathischen Arthritis (SIJA) und neu seit 2021 Jahr als einzige Therapie in der Schweiz zur Behandlung des adulten Still Syndroms (AOSD) zugelassen. Der IL-6 Rezeptor Antagonist Tocilizumab (TCZ) ist bisher nur für die Behandlung der SIJA zugelassen. Der humane IL-1 Rezeptorantagonist Anakinra ist in der CH nicht zugelassen.
In einer kleinen randomisierten Studie wurde TCZ 8 mg/kg KG alle 2 Wochen gegenüber Placebo bei steroidrefraktären Patienten mit AOSD getestet. Obwohl der primäre Endpunkt, die ACR50 Response, nicht erreicht wurde, zeigen sich in den sekundären Endpunkten (z.B. Möglichkeit der Steroidreduktion) Hinweise auf ein Ansprechen. Probleme waren die hohen Ansprechraten auf Placebo, möglicherweise auch der fortgeschrittene Krankheitsverlauf und die kleine Zahl von 27 Patienten 9.
Auch Canakinumab wurde in einer randomisierten Phase II Studie mit 41 Patienten gegen Placebo verglichen. Eingeschlossen wurden wiederum Patienten mit aktiver Erkrankung und einem DAS>5 und aktiver Arthritis. Während der Patientenrekrutierung für die Studie wurde Canakinumab von der EMA für die Behandlung von AOSD zugelassen. Aufgrund dessen konnten nicht mehr die geplanten Patientenzahlen rekrutiert werden, und die Studie wurde mit den bis zur Zulassung rekrutierten Patienten zu Ende geführt. Auch hier wurde der primäre Endpunkt (Reduktion des DAS um 1.2 Punkte) nicht erreicht. Obwohl die meisten sekundären Endpunkte in den mit Canakinumab behandelten Patienten deutlich häufiger als bei den Placebo behandelten Patienten erreicht wurden, waren die Unterschiede nicht statistisch signifikant. Auch hier handelte es sich um vorbehandelte Patienten mit etablierter Arthritis.10
Kohorten Daten stützen die Wirksamkeit von Canakinumab in der Behandlung des AOSD 11. In dieser retrospektiven multizenter Analyse zeigte sich bei 78% von 50 eingeschlossenen Patienten mit vorbehandelter, refraktärer Erkrankung ein gutes Ansprechen auf Canakinumab.
Neu in der klinischen Prüfung ist der monoklonale Antikörper Tadekinig, der gegen IL-18 gerichtet ist. Erste Studien zeigen eine gute Verträglichkeit und geben erste Hinweise auf eine mögliche Wirksamkeit 8.
Für Patienten mit etablierter chronischer Arthritis werden oft synthetische DMARDs, vor allem Methotrexat, eingesetzt. Alternativ kann auch die Gabe eines TNF-alpha Antagonisten sinnvoll sein.
Ausgehend von einem bi-phasischen Verlauf wird postuliert, dass eine frühe intensive antiinflammtorische Therapie den Übergang von innater Immunantwort (Autoinflammation), zur adaptiven Immunantwort (Autoimmunerkrankung) verhindern könnte und so der Entwicklung einer chronischen möglicherweise destruktiv verlaufenden Gelenkerkrankung vorbeugen könnte 9. Die Yamaguchi Klassifikationskriterien tragen diesem bi-phasischen Krankheitsverlauf Rechnung, da sie die Diagnose des AOSD in der frühen inflammatorischen Phase auch ohne Vorhandensein einer Arthritis ermöglichen.
Emapalumab, ein Antikörper (Ak) gegen INF-γ, hat in einer Pilotstudie eine Wirksamkeit gegen das HLH bei Patienten mit systemischer juveniler idiopathischer Arthritis sowie auch in Fallberichten bei AOSD gezeigt 10.
Zusammenfassung
Morbus Still ist eine typische ‘Rare Disease’. Damit geht einher, dass Evidenz über Studien mit grösseren Patientenzahlen nur sehr schwierig zu generieren ist. In der Diagnostik bedarf es angesichts des Fehlens spezifischer Marker eines sorgfältigen Ausschlusses alternativer Diagnosen. Es gibt zunehmend Hinweise dafür, dass eine frühe intensive, spezifische antiinflammatorische Therapie das Risiko für die Entwicklung einer chronisch destruktiven Arthropathie senken kann. Deswegen wird übereinstimmend empfohlen, Anti-IL-1 oder IL-6 gerichtete Therapien früh einzusetzen. IL-18 ist ein weiteres interessantes therapeutisches Ziel, das zurzeit geprüft wird. Bei bestehender chronischer Arthropathie kann MTX oder auch ein TNFi eingesetzt werden. Der Stellenwert neuerer Therapieansätze (JAKi, IL-17 Antagonisten etc.) ist noch unklar.
Die Etablierung von Kohorten mit AOSD Patienten ermöglicht die Generierung weiterer Evidenz und sollte breit gefördert werden.
Referenzen
- Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-onset Still’s disease. Autoimmunity Reviews. 1. Juli 2014;13(7):708–22.
- Foell D, Wittkowski H, Kessel C, Lüken A, Weinhage T, Varga G, u. a. Proinflammatory S100A12 can activate human monocytes via Toll-like receptor 4. Am J Respir Crit Care Med. 15. Juni 2013;187(12):1324–34.
- Kessel C, Lippitz K, Weinhage T, Hinze C, Wittkowski H, Holzinger D, u. a. Proinflammatory Cytokine Environments Can Drive Interleukin-17 Overexpression by γ/δ T Cells in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. Juli 2017;69(7):1480–94.
- Fautrel B, Le Moël G, Saint-Marcoux B, Taupin P, Vignes S, Rozenberg S, u. a. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J Rheumatol. Februar 2001;28(2):322–9.
- Szekanecz Z, McInnes IB, Schett G, Szamosi S, Benkő S, Szűcs G. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat Rev Rheumatol. Oktober 2021;17(10):585–95.
- Tarp S, Amarilyo G, Foeldvari I, Christensen R, Woo JMP, Cohen N, u. a. Efficacy and safety of biological agents for systemic juvenile idiopathic arthritis: a systematic review and meta-analysis of randomized trials. Rheumatology (Oxford). April 2016;55(4):669–79.
- Kaneko Y, Kameda H, Ikeda K, Ishii T, Murakami K, Takamatsu H, u. a. Tocilizumab in patients with adult-onset still’s disease refractory to glucocorticoid treatment: a randomised, double-blind, placebo-controlled phase III trial. Ann Rheum Dis. Dezember 2018;77(12):1720–9.
- Gabay C, Fautrel B, Rech J, Spertini F, Feist E, Kötter I, u. a. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann Rheum Dis. Juni 2018;77(6):840–7.
- Föll D, Wittkowski H, Hinze C. Das Still-Syndrom als biphasische Erkrankung. Z Rheumatol. 1. September 2020;79(7):639–48.
- Gabr JB, Liu E, Mian S, Pillittere J, Bonilla E, Banki K, u. a. Successful treatment of secondary macrophage activation syndrome with emapalumab in a patient with newly diagnosed adult-onset Still’s disease: case report and review of the literature. Ann Transl Med. Juli 2020;8(14):887.
