Muskuloskelettale Beteiligung bei Akromegalie
Einleitung
Die Akromegalie gehört in der allgemeinmedizinischen Praxis sowie im regulären Krankenhausbetrieb zu den seltenen Erkrankungen. Ursache ist die Überproduktion des Hormons Somatotropin, meist als Folge eines autonomen Adenoms der (Adeno-) Hypophyse. Der Somatotropin-Überschuss stimuliert nach dem Abschluss des Knochenlängenwachstums, also im Erwachsenenalter, die Grössenzunahme von Akren und inneren Organen (Akro- und Viszeromegalie). Subjektiv sind die Betroffenen in bis zu 70% durch muskuloskelettale Beschwerden beeinträchtigt. Klinisch finden sich gehäuft drei Manifestationen: periphere Arthropathie, Pathologien an der Lendenwirbelsäule, welche in fortgeschrittenen Stadien kaum von einer üblichen Arthrose zu unterscheiden sind, sowie ein Karpaltunnelsyndrom. An den Sakroiliakalgelenken sind in bis zu 30% Folgen der STH-Überproduktion erkennbar, zumindest in der bildgebenden Diagnostik.
Akromegalie: Definition und Epidemiologie
Der Begriff Akromegalie beschreibt einen unkontrollierten Wachstumsprozess der Akren, hervorgerufen durch die Überproduktion des hypophysären Hormons Somatotropin (STH). Nur Erwachsene können das Krankheitsbild entwickeln, während der Wachstumsphase führt ein STH-Exzess hingegen zum (zerebralen) Gigantismus. Von prognostischer Bedeutung ist der Umstand, dass bei Akromegalie nicht allein die Akren, also Hände, Füsse und Kopf, sondern ebenso innere Organe betroffen sind (Viszeromegalie). Die Erkrankung manifestiert sich selten (Prävalenz ca. 0,3/100.000 Einwohner jährlich), Erstmanifestationsalter ist meist die 4.–5. Lebensdekade.
Pathogenese
Somatotropin wird in der Adenohypophyse synthetisiert, die Produktion variiert altersund tagesabhängig. So besteht nächtlich eine vermehrte Sekretion, vor allem während der Pubertät. Hypoglykämie stimuliert die Freisetzung, Nahrungsaufnahme hingegen wirkt inhibitorisch. Die Aktivität der Hypophyse unterliegt hypothalamischer Kontrolle. Somatoliberin (Growth Hormone-Releasing Hormone – GH-RH) wirkt agonistisch, Somatostatin hingegen antagonistisch auf die STH-Produktion. Die peripheren STH-Effekte werden durch Somatomedin C (Insulin-like Growth Factor-1 – IGF-1) vermittelt 1. Bei der Akromegalie ist die entkoppelte STH-Produktion fast ausnahmslos auf ein hormonaktives Hypophysenadenom zurückzuführen 2. In sehr seltenen Fällen wurde ätiologisch eine entweder hypothalamische oder ektope GH-RH-Überproduktion beschrieben 3.
Klinisches Bild in der Übersicht
Wie zahlreiche andere Erkrankungen manifestiert sich die Akromegalie initial eher unspezifisch. Typische Frühsymptome umfassen Schwitzen und Kopf- oder Gelenkschmerzen. Angesichts der geringen Prävalenz ist es kaum überraschend, dass in einer solchen Situation nur äusserst selten ein hypophysärer Hormonexzess als Ursache bedacht wird. Auch arterielle Hypertonie sowie konzentrische Hypertrophie des linken Ventrikels sind diagnostisch kaum richtungsweisend. Nach vielen Jahren Krankheitsdauer hingegen präsentieren Betroffene meist typische physiognomische Auffälligkeiten, welche nunmehr fast ausschliesslich bei der Akromegalie vorkommen (Vergröberung der Gesichtszüge, Zunahme der Schuhgrösse). In diesem Stadium hat die permanent vermehrte Bildung von IGF-1 jedoch meist schon irreversible Strukturveränderungen am Bewegungsapparat sowie inneren Organen verursacht.
Muskuloskelettale Beteiligung
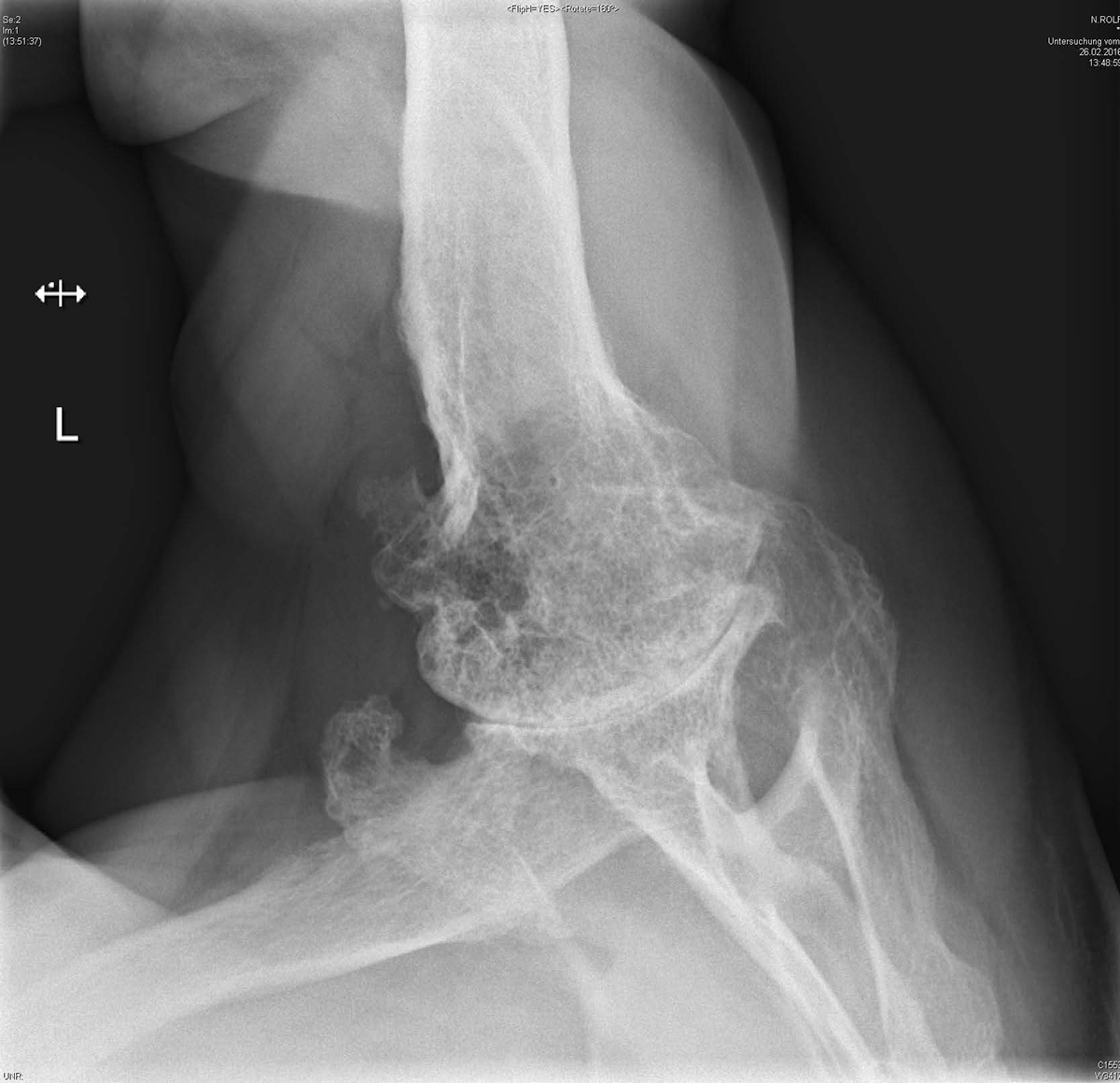
Bis zu 70% aller Betroffenen entwickeln im Verlauf der Erkrankung muskuloskelettale Komplikationen. Typische Symptome umfassen Schmerzen an Schultern, Hüften/Knien (grössere Gelenke) sowie am unteren Rücken. Ein Karpaltunnelsyndrom manifestiert sich bei bis zu 50% der Patient:innen 4. Schmerzen im LWS-Bereich treten ebenfalls bei ca. 50% aller Betroffenen auf 4–6. Die Knochenstabilität ist oft beeinträchtigt, sowohl eine Osteoporose, als auch erhöhte Knochendichtewerte wurden beschrieben 7, 8. Die muskuloskelettale Beteiligung resultiert überwiegend aus den anabolen Effekten der STH-/IGF-1-Achse. Die Aktivität von perichondralen Zellen nimmt unter dem Einfluss von IGF-1 zu 9. Knorpelverdickungen und Strukturveränderungen am Gelenkbandapparat verursachen artikuläre Mikrotraumata sowie die (reaktive) Bildung von Osteophyten. Letztere finden sich auch an der Wirbelsäule. In fortgeschrittenen Fällen ist kaum eine Unterscheidung von üblichen degenerativen Gelenkschäden möglich (Abbildung 1).
Die klinischen Symptome infolge der Knochenveränderungen sind meist einem von drei Komplexen zuzuordnen: (I) Arthropathie grösserer Gelenke, (II) lumbale Schmerzen / Funktionsstörungen bzw. ein LWSSyndrom sowie (III) das Karpaltunnelsyndrom (Abbildung 2).
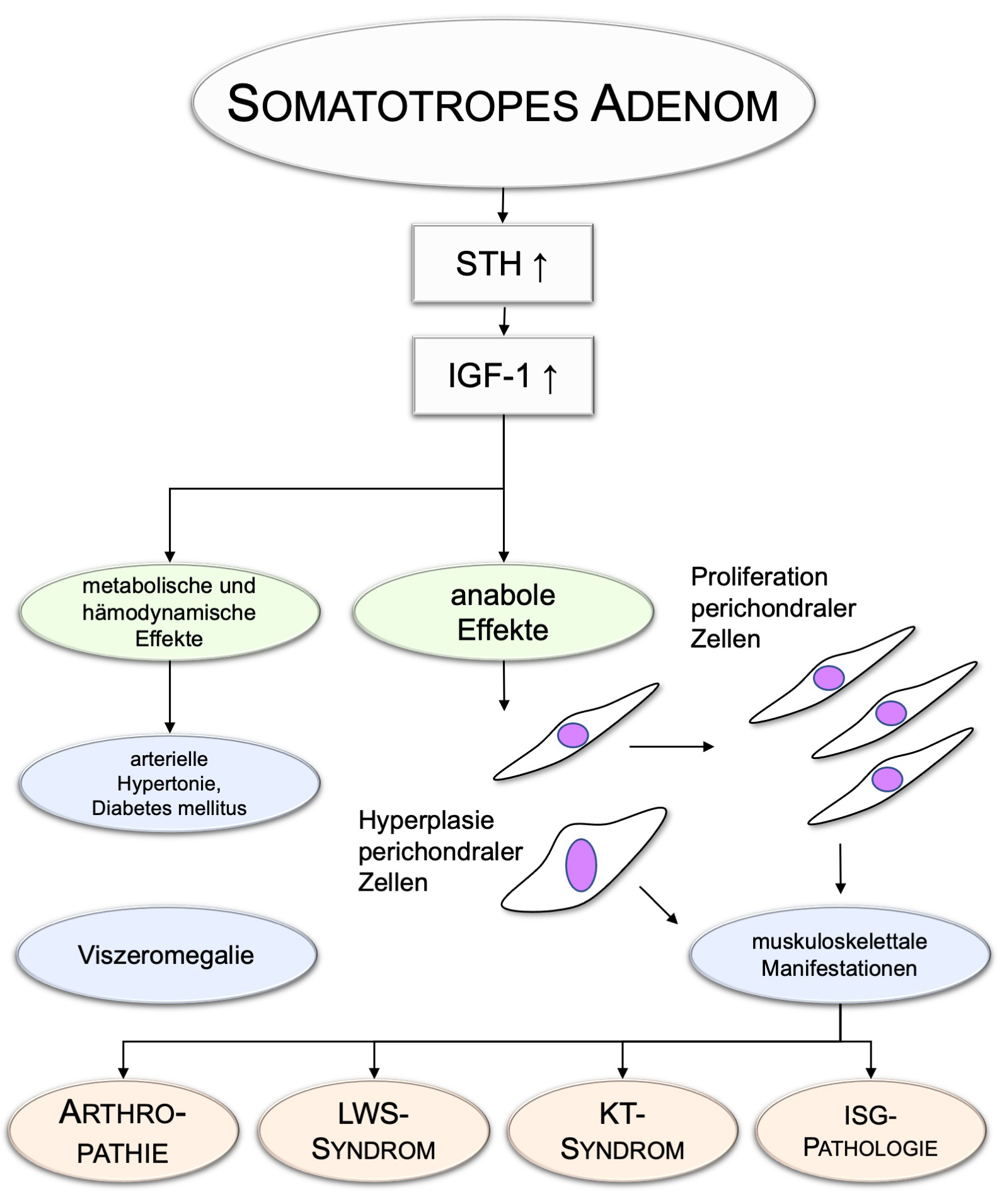
Letzteres ist auch auf modulatorische Effekte von Somatotropin auf die Aktivität bestimmter Natriumkanäle zurückzuführen
(zellulärer Volumenzuwachs, vor allem am Nervus medianus 4, 10). Die anabolen Wirkungen des Somatotropins gehen an der Lendenwirbelsäule mit ossärer Hypertrophie und der Expansion der Zwischenwirbelscheiben einher. In schweren Fällen entsteht an der LWS eine so benannte diffuse skelettale Hyperostose 11. Im Jahr 2019 berichteten Ugur und Kolleg:innen ausserdem über strukturelle Veränderungen in den Sakroiliakalgelenken von 33 Akromegalie-Patient:innen 12. In der konventionellen Röntgendiagnostik ergaben sich dabei Auffälligkeiten in ca. 36%, magnetresonanztomographisch hingegen zeigten sich in etwa 12% suspekte Befunde. Daher sollte vor allem im Rahmen der Abklärung des Verdachts auf eine seronegative Spondylarthropathie an die Akromegalie gedacht werden, sofern sich in den ISG-Gelenken pathologische Befunde zeigen.
Diagnostik
Üblicherweise finden sich bei Akromegalie signifikant erhöhtes STH- und IGF-1-Konzentrationen im Serum. Typisch ist zudem der ausbleibende Abfall der Somatotropinkonzentration trotz Zufuhr von Glukose. Lassen Klinik und Laborbefunde eine Akromegalie vermuten, ist bildgebende Diagnostik unverzichtbar. Mittels MRT sind Mikrooder Makroadenome (< vs. ≥1 cm 13) des Hypophysenvorderlappens meist sicher diagnostizierbar. Spezialuntersuchungen, wie etwa eine Octreotid-Szintigraphie werden selten benötigt, da eine hypothalamische oder ektope GH-RH-Überproduktion
kaum je ursächlich sind.
Behandlungsoptionen
In den weitaus meisten Fällen ist die operative Entfernung des STH-produzierenden Hypophysenadenoms (vorzugsweise transsphenoidal) Therapie der Wahl. Im Falle von grösseren Adenomen (>1 cm – Makroadenomen) ist die vollständige Entfernung mitunter schwierig. Allerdings erhöht bereits die teilweise Entfernung des Adenomgewebes die Erfolgschancen einer nachfolgenden Pharmakotherapie. Dopaminantagonisten wie z.B. Cabergolin kommen zum Einsatz, wenn trotz OP noch ein relevanter STHExzess besteht. Eine weitere Medikamentengruppe sind Somatostatin-Analoga, welche auch kombiniert mit Dopaminantagonisten angewendet werden können. Sofern chirurgisches Vorgehen und medikamentöse Therapie versagen, sind mitunter strahlentherapeutische Verfahren indiziert 13.
Zusammenfassung
- Die Akromegalie ist eine seltene Erkrankung.
- Ursache ist in den meisten Fällen ein somatotropes Adenom der Adenohypophyse.
- Die anabolen Effekte von STH verursachen perichondrale und ossäre Hyperplasien.
- Potentielle Manifestationen am Bewegungsapparat umfassen periphere Arthropathie, Pathologien an der Lendenwirbelsäule sowie ein Karpaltunnelsyndrom. Radiologisch finden sich an den Sakroiliakalgelenke in bis zu 30% auffällige Befunde.
- Therapie der Wahl ist die operative Adenomentfernung, ggf. ergänzt um medikamentöse Massnahmen bei unzureichender Resektion.
Referenzen
- O. O. Adigun, M. Nguyen, T. J. Fox, und C. Anastasopoulou, „Acromegaly“, in StatPearls, Treasure Island (FL): StatPearls Publishing, 2021. Zugegriffen: Nov. 08, 2021. [Online]. Verfügbar unter: http://www.ncbi.nlm.nih.gov/books/NBK431086/
- R. Dineen, P. M. Stewart, und M. Sherlock, „Acromegaly“, QJM Mon. J. Assoc. Physicians, Bd. 110, Nr. 7, S. 411–420, Juli 2017, doi: 10.1093/qjmed/hcw004.
- S. Melmed, „Acromegaly pathogenesis and treatment“, J. Clin. Invest., Bd. 119, Nr. 11, S. 3189– 3202, Nov. 2009, doi: 10.1172/JCI39375.
- Z. Killinger u. a., „Arthropathy in acromegaly“, Rheum. Dis. Clin. North Am., Bd. 36, Nr. 4, S. 713– 720, Nov. 2010, doi: 10.1016/j.rdc.2010.09.004.
- L. C. Detenbeck, H. A. Tressler, J. D. O’Duffy, und R. V. Randall, „Peripheral joint manifestations of acromegaly“, Clin. Orthop., Nr. 91, S. 119–127, Apr. 1973.
- M. Podgorski, B. Robinson, A. Weissberger, J. Stiel, S. Wang, und P. M. Brooks, „Articular manifestations of acromegaly“, Aust. N. Z. J. Med., Bd. 18, Nr. 1, S. 28–35, Feb. 1988.
- M. J. E. Wassenaar u. a., „Clinical osteoarthritis predicts physical and psychological QoL in acromegaly patients“, Growth Horm. IGF Res. Off. J. Growth Horm. Res. Soc. Int. IGF Res. Soc., Bd. 20, Nr. 3, S. 226–233, Juni 2010, doi: 10.1016/j.ghir.2010.02.003.
- M. Bolanowski, J. Daroszewski, M. Medraś, und B. Zadrozna-Sliwka, „Bone mineral density and turnover in patients with acromegaly in relation to sex, disease activity, and gonadal function“, J. Bone Miner. Metab., Bd. 24, Nr. 1, S. 72–78, 2006, doi: 10.1007/s00774-005-0649-9.
- K. Okazaki u. a., „Expression of insulin-like growth factor I messenger ribonucleic acid in developing osteophytes in murine experimental osteoarthritis and in rats inoculated with growth hormonesecreting tumor“, Endocrinology, Bd. 140, Nr. 10, S. 4821–4830, Okt. 1999, doi: 10.1210/endo.140.10.7053.
- P. Kamenicky u. a., „Body fluid expansion in acromegaly is related to enhanced epithelial sodium channel (ENaC) activity“, J. Clin. Endocrinol. Metab., Bd. 96, Nr. 7, S. 2127–2135, Juli 2011, doi: 10.1210/jc.2011-0078.
- L. Altomonte u. a., „Growth hormone secretion in diffuse idiopathic skeletal hyperostosis“, Ann. Ital. Med. Interna Organo Uff. Della Soc. Ital. Med. Interna, Bd. 7, Nr. 1, S. 30–33, März 1992.
- K. Ugur, A. Karatas, B. Oz, H. Artas, S. Aydin, und S. S. Koca, „Imaging of sacroiliac joints in patients with acromegaly“, Sci. Rep., Bd. 9, Nr. 1, S. 11645, Aug. 2019, doi: 10.1038/s41598-019-48250-w.
- L. Katznelson u. a., „Acromegaly: an endocrine society clinical practice guideline“, J. Clin. Endocrinol. Metab., Bd. 99, Nr. 11, S. 3933–3951, Nov. 2014, doi: 10.1210/jc.2014-2700.
