Hypermobility Spectrum Disorder (ab 2017) (HSD)
Spektrum der Erkrankungen, die einhergehen mit einer Hypermobilität
Terminologie
Die Hypermobilität gehört zur Gruppe der hereditären Bindegewebserkrankungen. 2017 wurden die internationalen Kriterien zur Differenzierung dieser verschiedenen Erkrankungen mit einer Hypermobilität neu erarbeitet und definiert. Die Bezeichnung Hypermobilitätssyndrom wurde ersetzt durch a.) Das hypermobile Ehlers-Danlos-Syndrom und b.) Hypermobility Spectrum Disorder (HSD). Fehlen Hinweise auf ein eigentliches Ehlers-Danlos-Syndrom bei einer Hypermobilität, so spricht man von einer Hypermobility Spectrum Disorder. In diese Gruppe gehört das früher bezeichnete Hypermobilitätssyndrom/Hyperlaxizitätssyndrom.
Epidemiologie
Eine Hypermobilität (generalisiert oder lokal) findet sich in 10%–20% der gesunden Bevölkerung. Vermehrt betroffen sind, Kinder, Jugendliche, Frauen sowie die asiatische und westafrikanische Bevölkerungsgruppe. Eine Hypermobilität (HSD) mit Funktionseinschränkungen und Schmerzen ist deutlich seltener, das Auftreten liegt bei 0.1% in der Bevölkerung.
Ätiologie/Pathogenese
Eine genetische Disposition ist vorhanden, da sich gehäuft Fälle mit Hypermobilität in Familien/Verwandtschaften finden. Das Ausmass der Hypermobilität und die Symptome können aber innerhalb einer Familie sehr unterschiedlich sein. Der genaue genetische Defekt ist noch unbekannt.
Beim eigentlichen Ehlers-Danlos-Syndrom und beim Marfan-Syndrom sind die genetisch bedingten Störungen des Kollagens und der Fibrillen im Bindegewebe bekannt und können bestimmt werden.
Klinische Manifestationen
Die Patienten klagen über wechselnde Schmerzen an Gelenken und Muskeln, die häufig bei der Erstkonsultation nicht klar zugeordnet werden können. Je nach Alter treten Distorsionen (v.a. OSG) und Subluxationen bis Luxationen (Schultern, Handgelenke, Knie, OSG) treten bei Hypermobilität gehäuft auf. In der Anamnese muss gezielt nachgefragt werden, in welchen Gelenken es bereits im Kindes- und Jugendalter zu Distorsionen kam und welche überdurchschnittlichen Bewegungen (z.B. ein Spagat) schon damals möglich waren. Die Mehrheit der Patienten berichtet, dass sie im Turnunterricht immer zu den besten gehörten, dies ohne grosse Anstrengung.
Weitere Symptome, die mit einer HSD einhergehen können, sind:
- Schmerzen, lokalisiert bis generalisiert
- schlechte Propriozeption
- Koordination
- Gleichgewichtsstörungen
- allgemeine Müdigkeit
- Störungen des autonomen Nervensystems
- kardio-vaskuläre Störungen
- gastro-intestinale Störungen
- urogenitale Störungen
- Angstzustände, Depression und Phobien
Auch die Haut ist oft überdehnbar, es finden sich Striae und papierdünne Narbenbildungen. In etwa einem Drittel der Fälle ist der Körperhabitus marfanoid.
Gelenksschmerzen können sehr gering bis sehr stark sein. Sie variieren häufig und verleiten zu Fehldiagnosen (z.B. entzündliche Gelenkserkrankungen, lokalisiertes oder generalisiertes Schmerzsyndrom).
Diagnostik
Die Diagnose einer HDS wird anhand einer detaillierten Anamnese und der körperlichen Untersuchung (Beighton-Kriterien, Abb. 1 und 2) gestellt. Genetische, laborchemische und bildgebende Abklärungen sind für die Diagnose einer HDS nicht nötig.
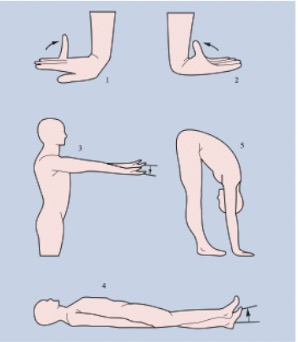
Quelle: Michel BA, Brühlmann P, Meier B: Rheumatologie: Klinische Untersuchung. Rheuma Schweiz 2020:56
- Kleinfingerextension parallel zum Vorderarm
- Daumenadduktion zum Vorderarm
- Hyperextension Ellbogen > 10°
- Hyperextension Knie > 10°
- In Inklination flache Hände auf BodenBeighton Score total 9 Punkte:
Teste 1–4 bei Doppelseitigkeit je 2 Punkte
Test 5 ergibt 1 PunktGeneralisierte Hypermobilität bei Score >=4/9
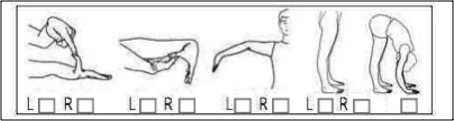
Quelle: https://www.ehlers-danlos.com/wp-content/uploads/heds-diagnostic-checklist-german.pdf)
- > 6 vorpubertäre Kinder und Jugendliche
- > 5 Männer und Frauen von der Pubertät bis 50 Jahre
- > 4 Männer und Frauen über 50 Jahre
Eine klare Eingrenzung der Diagnose anhand der Anamnese ist häufig schwierig. Differentialdiagnostisch muss an ein Marfan-Syndrom, ein Loeys-Dietz Syndrom und ein Ehlers-Danlos Syndrom gedacht werden.
Marfan Syndrom (MS) und Loeys-Dietz-Syndrom (LDS)
Das MS tritt mit einer Prävalenz von 1–5:10 000 selten, das LDS mit 1:100.000 sehr selten auf.
Das MS und LDS sind Multisystemerkrankungen, welche in ihrer Organverteilung, dem Alter der Erstmanifestation und der Schwere des Verlaufs sehr variabel sind (Tabelle 1).
Tabelle 1: Manifestationen bei Marfan- und Loeys-Dietz-Syndrom 3
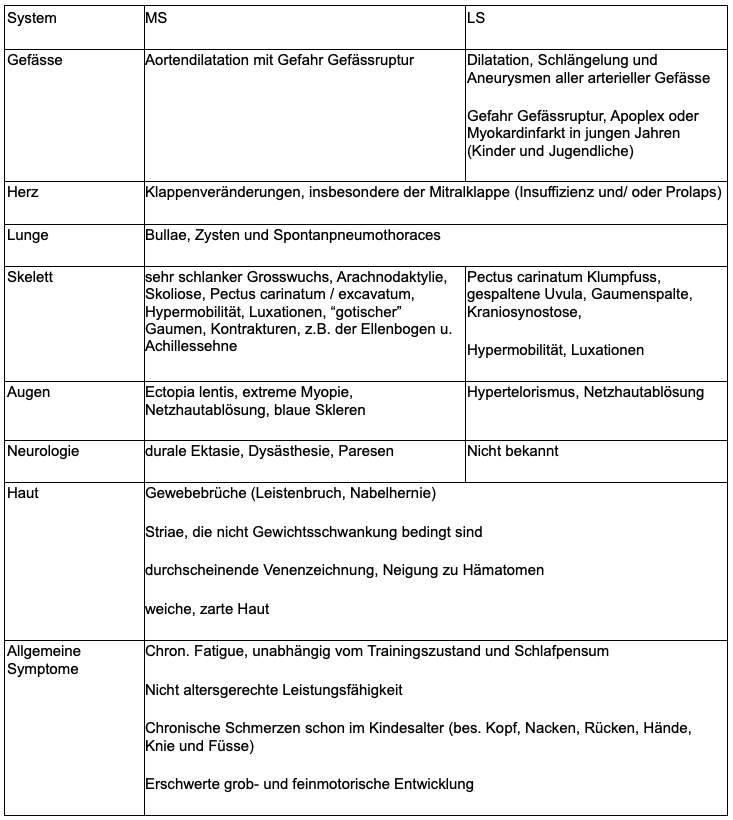
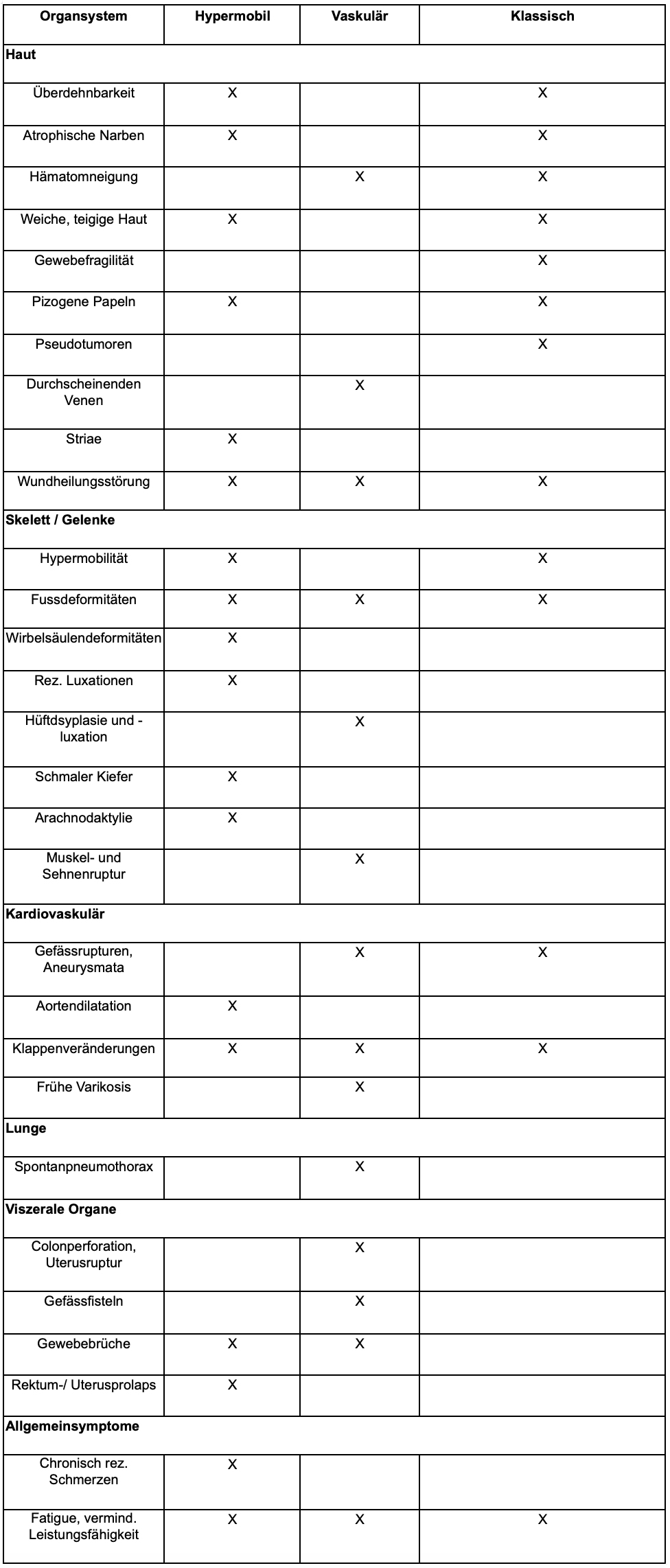
So weit wie möglich ist durch die Anamnese und die körperliche Untersuchung eine Abgrenzung zu einem Ehlers-Danlos-Syndrom (EDS) mit seinen 3 Varianten (hypermobil, vaskulär, klassisch) nötig (Tabelle 2).
Da das hypermobile EDS noch nicht molekulargenetisch verifiziert werden kann, erfolgt die Diagnose anhand einer Checkliste, welche die Hypermobilität, andere Manifestationen an Haut, Herz, viszeralen Organen, die Schmerzanamnese, die Familienanamnese und den Ausschluss rheumatologischer oder anderer Bindegewebesyndrome einbezieht.
Tabelle 2: Manifestationen der 3 Varianten des Ehlers-Danlos-Syndroms 4:
Therapie
Die Behandlung orientiert sich nach den Funktionseinschränkungen und den Schmerzen im Alltag (Ergonomie im Beruf, Haushalt, Freitzeitaktivitäten, Sport). Die physikalische Therapie muss individuell angepasst sein und berücksichtigt die Lokalisation, die Dauer und das Ausmass der Funktionseinschränkungen und Schmerzen zum Zeitpunkt der Untersuchung und anhand der Anamnese (insbesondere Erkrankungsdauer, Verlauf, Erfahrungen). Die muskuläre Stabilisierung der Gelenke und der Wirbelsäule stehen im Vordergrund.
Idealerweise werden Patienten mit einer HSD durch ein erfahrendes Team (Hausarzt, Facharzt, Physiotherapie, Ergotherapie, klinische Psychologie) über längere Zeit begleitet und behandelt. Die Patienten lernen mit ihren chronischen Beschwerden umzugehen mit dem Ziel ein möglichst normales Leben führen zu können.
In Situationen einer Schmerzexazerbation kann der Einsatz von Analgetika, NSAR und Antidepressiva hilfreich sein. Vermeiden sollte man eine Medikation mit Opiaten und Muskelrelaxantien.
Verlauf/Prognose
Mit zunehmendem Alter werden die Bänder allgemein fester und damit die Gelenke weniger beweglich, so dass Komplikationen wie Sub- und Luxationen nicht mehr bis seltener auftreten.
Weiterführende Links
Abklärung durch ein spezialisiertes Zentrum:
Zentrum für seltene Krankheiten Zürich an der Universitätsklinik Balgrist (https://www.zentrumseltenekrankheiten.ch). Beteiligt an der Sprechstunde sind:
Universitäts-Kinderspital Zürich, UniversitätsSpital Zürich, Universitätsklinik Balgrist sowie Institut für medizinische Genetik der Universität Zürich.
Ehlers-Danlos-Society (https://www.ehlers-danlos.com)
- Broschüren: https://www.ehlers-danlos.com/brochures/
- Kontaktstelle in der Schweiz: https://www.ehlers-danlos.com/healthcare-professionals-directory-switzerland/
Übersicht:
Hakim AJ, Keer R, Grahame R. Hypermobility, Fibromyalgia and Chronic Pain, Churchill Livingstone, Elsevier, 2010.
Referenzen
- Michel BA, Brühlmann P, Meier B: Rheumatologie: Klinische Untersuchung. Rheuma Schweiz 2020:56
- https://www.ehlers-danlos.com/wp-content/uploads/heds-diagnostic-checklist-german.pdf
- Link B, Rohrbach M. Hypermobilitäts-Syndrom, hereditäre Bindegewebserkrankungen. Zeitschrift Rheuma Schweiz 2021;3:15
- Link B, Rohrbach M. Hypermobilitäts-Syndrom, hereditäre Bindegewebserkrankungen. Zeitschrift Rheuma Schweiz 2021;3:15
