Einleitung
Autoinflammatorische Erkrankungen (AID) bilden eine heterogene Gruppe von seltenen Erkrankungen (rare diseases/orphan diseases), bei denen es zu einer systemischen episodischen oder dauerhaften Entzündungsreaktion kommt1. Häufig verwendete Synonyme sind periodische Fiebersyndrome oder hereditäre Fiebersyndrome. Wichtig ist hierbei zu bedenken, dass die Fiebersymptomatik fehlen kann. Im englischen Sprachgebrauch wird meistens der Begriff autoinflammatory disease (AID) verwendet. Der Terminus Autoinflammation wurde Ende der 1990er Jahre eingeführt2. Initial wurde die Autoinflammation in Abgrenzung zur Autoimmunität gesehen und hat Erkrankungen zusammengefasst, bei denen Entzündung durch Effektorzellen des angeborenen Immunsystems vermittelt wurde und in Abgrenzung zu Autoimmunerkrankungen antigenspezifische T-Zellen und Antikörper keine zentrale Rolle spielen. Heutzutage weiss man, dass es zwischen den Entitäten zunehmend Überlappungen geben kann3.
Die Häufigkeiten von AID variieren; zum einen zwischen den verschiedenen AID, aber auch in regionaler und ethnischer Hinsicht. Von einer hohen Dunkelziffer ist auszugehen. In den letzten Jahren hat das Wissen im Bereich von monogenetischen und polygenetischen AID kontinuierlich zugenommen. Durch die Etablierung von genetischen Testverfahren wie Next-Generation-Sequencing (NGS), wächst das Spektrum an beschriebenen AID auf molekularer und genetischer Ebene fortlaufend4. Die Diagnosestellung einer AID ist oft herausfordernd. So können Phänotypen nicht immer einem Genotyp zugeordnet werden, auch können bekannte Phänotypen in ihrer Symptomatik variieren5, 6. Viele erkrankte Kinder und Jugendliche haben daher oft einen langen und teils belastenden Weg bis zur Diagnose7,8, wie auch die beiliegende Kasuistik illustriert.
Unbehandelt gehen viele AID mit erheblicher Morbidität, einem erhöhten Mortalitätsrisiko sowie eingeschränkter gesundheitsbezogener Lebensqualität einher; sie haben negative Auswirkungen auf das Sozialleben der Betroffenen und der gesamten Familie und bergen das Risiko für psychische Komorbiditäten9–13. Da mit Diagnosestellung, insbesondere bei Interleukin (IL)-1 vermittelten AID, eine wirksame Therapie eingeleitet werden kann, möchten wir im Folgenden auf einige AID steckbriefartig näher eingehen und klinische und laborchemische Charakteristika benennen, die an eine AID denken lassen sollten.
Kasuistik
Im Rahmen der Erstvorstellung berichten die Eltern von ihrem 7 Jahre alten Sohn. Seit dem 15. Lebensmonat käme es zu rezidivierenden, 2–3 Tage andauernden, nicht juckenden Hautausschlägen. Ab dem 2. Lebensjahr sei aufgefallen, dass zeitgleich oft subfebrile bis febrile Temperaturen bestünden. Oft sei es begleitend zu geröteten Augen und Gehverweigerung gekommen. In den Episoden hätte sich ein mässig erhöhtes C-reaktives Protein (CRP) gezeigt, so dass die Symptomatik infektassoziiert gewertet worden sei. Im Kindergarten sei aufgefallen, dass der Sohn weniger belastbar gewesen wäre als Gleichaltrige. Im letzten Jahr würden die Hautausschläge, das Fieber, die geröteten Augen und die Gelenkschmerzen zusammen mit Kopfschmerzen vermehrt auftreten, und es käme zu regelmässigen Fehlzeiten in der Schule über 3–4 Tage. Es erfolgte daher eine Vorstellung beim Kinderarzt mit Zuweisung in die Kinderrheumatologie. Zwei Tage vor der Vorstellung hätte ein typischer Krankheitsschub mit Fieber, Hautausschlag, Arthralgien und Konjunktivitis geendet. Die Familie hat ein Foto des Hautausschlags dabei (Abbildung 1).
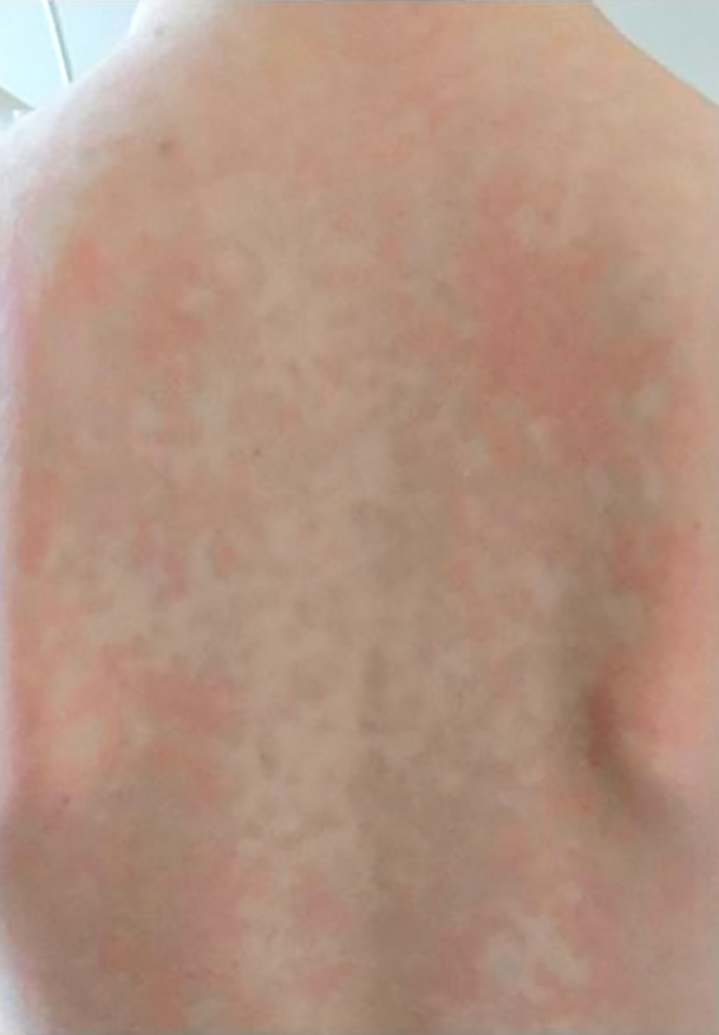
Der körperliche Status zeigt sich bis auf eine Konjunktivitis unauffällig.
In der Laboruntersuchung sind die Entzündungswerte (CRP 80 mg/L, Blutsenkungsreaktion (BSG) 40 mm/h, Serum Amyloid A (SAA) 400 mg/l [Norm < 10]) erhöht, weitere Organfunktionsparameter sind unauffällig. In der Hochfrequenz-Audiometrie zeigt sich eine moderate Schallempfindungs-Schwerhörigkeit links. Es wird die Diagnose CAPS (moderater Phänotyp) basierend auf den Diagnosekriterien gestellt und eine Therapie mit einem zugelassenen IL-1 Inhibitor begonnen. Die Genetik zeigt eine pathologische Variante im NLRP3-Gen (p.Thr348Met). In der Verlaufskontrolle nach 6 Monaten berichteten die Eltern, dass es unter der Therapie zu keinen charakteristischen Krankheitsschüben mehr gekommen sei, allerdings sei noch zweimal nach Kälteexposition der Hautausschlag für 1–2 Tage aufgetreten. Der Sohn habe mehr Energie und könne nun bei körperlichen Aktivitäten ausdauernd mit den Spielkameraden mithalten. In der nachfolgenden Audiometrie zeigte sich nur noch eine diskrete linksseitige hochtonale Schwerhörigkeit. CRP und SAA waren im Normbereich.
Steckbriefe einiger ausgewählter AID
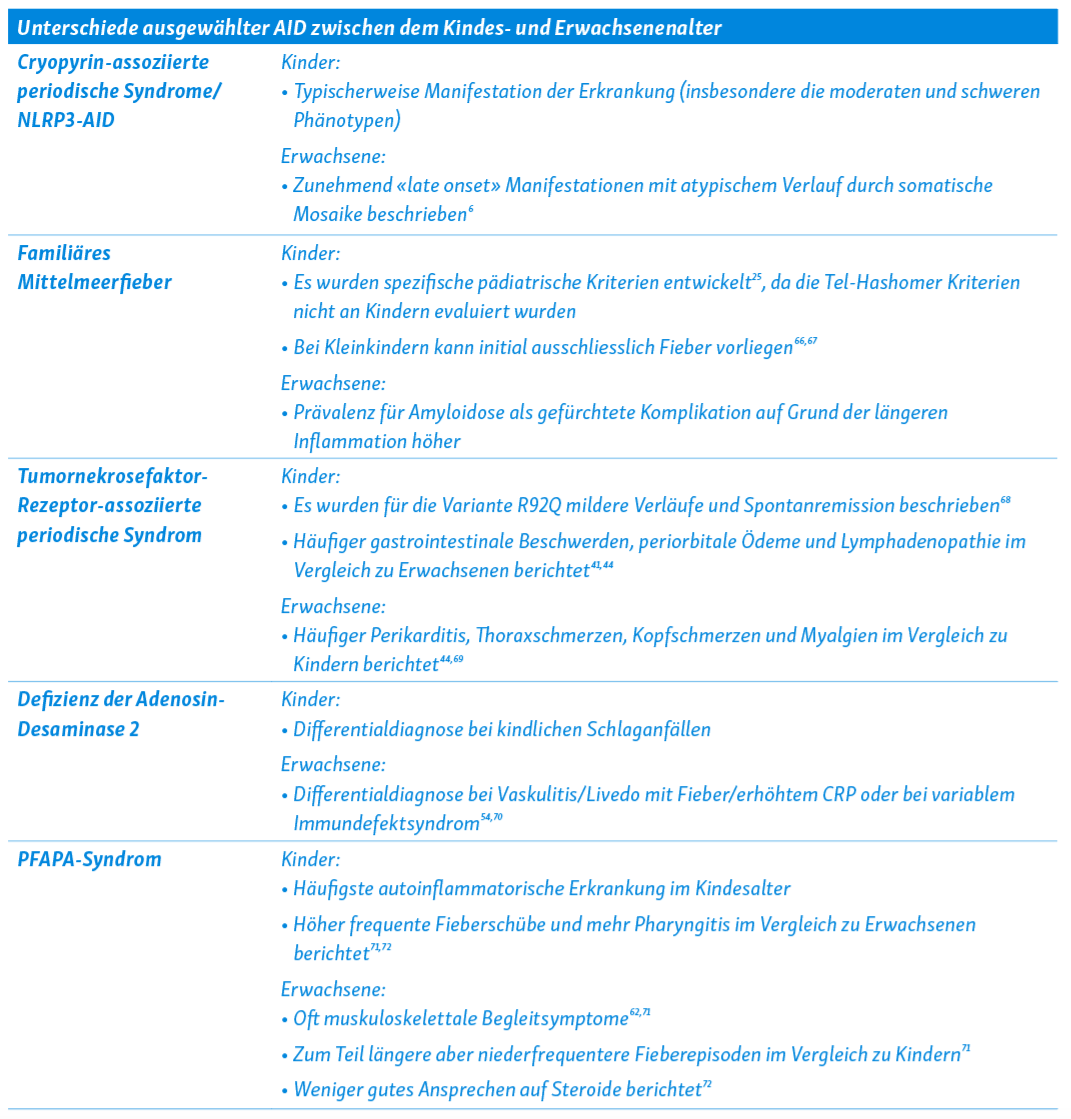
Im Folgenden finden sich kurze Streckbriefe zu einzelnen ausgewählten AID. Tabelle 1 fasst einige Unterschiede bei diesen Erkrankungen zwischen Kindern und Erwachsenen zusammen.
Cryopyrin-assoziierte periodische Syndrome (CAPS)
CAPS werden heute im Zuge der neuen Taxonomie als NLRP3-assoziierte AID (NLRP3-AID) bezeichnet14. Sie entsprechen einem Krankheitsspektrum mit Phänotypen verschiedener Schweregrade, zu denen die milden Phänotypen als Familial Cold Autoinflammatory Syndrome (FCAS) bekannt sind, moderate Phänotypen auch als Muckle-Wells-Syndrom (MWS) benannt werden und die schweren Phänotypen als Neonatal Onset Multisystem Inflammatory Disease (NOMID) oder Chronic Infantile Neurological Cutaneous and Articular Syndrome (CINCA) bezeichnet werden15. Bei den meisten CAPS-Patienten können pathogene gain-of-function-Varianten im NLPR3-Gen gefunden werden, die zu einer konstitutiven Aktivierung des NLRP3-Inflammasoms führen16. Während sich die schweren Phänotypen zumeist perinatal manifestieren, können leichte Phänotypen erst im Erwachsenenalter symptomatisch werden. Für alle drei Phänotypen sind Fieber, neutrophile Dermatitis und muskuloskelettale Beschwerden (Arthralgien, Arthritis) beschrieben; Innenohrschwerhörigkeit, ophthalmologische Entzündungen und Kopfschmerzen durch rezidivierende oder chronische aseptische Meningitis sind auf die moderaten und schweren Phänotypen beschränkt17, 18. Bei NOMID/CINCA zeigen sich zudem Auffälligkeiten im Skelettsystem. Patienten mit CAPS sollten mit einer IL-1 Inhibition behandelt werden19.
Familiäres Mittelmeerfieber (FMF)
Beim FMF handelt es sich um die häufigste weltweit vorkommende IL-1 vermittelte AID20. Für das FMF sind Veränderungen im MEFV-Gen erstmalig 1997 beschrieben worden21, 22. Während initial davon ausgegangen wurde, dass das FMF einem autosomal-rezessiven Erbgang folgt, geht man heutzutage von einer «Gendosis-Wirkung» aus23. Dies bedeutet, dass Patienten mit zwei klar pathogenen Varianten meist an einer schweren Verlaufsform mit erhöhtem Risiko für eine Amyloidose leiden, während FMF-Patienten mit nur einer Variante oder Träger niedrig pathogener Varianten an meist nur leicht ausgeprägter Symptomatik leiden24. Die Verdachtsdiagnose wird klinisch gestellt; Diagnosekriterien sind hierbei hilfreich. Kurze Fieberepisoden von 6–72 Stunden, welche von einer Serositis (sterile Peritonitis, Pleuritis) und/oder Arthritis begleitet werden, sind charakteristisch für FMF [25]. Weitere typische Symptome sind erysipelartige Erytheme, Myalgien oder Hodenschmerzen. Die Episodenfrequenz ist variabel, von wenigen Wochen bis einigen Monaten. Das Manifestationsalter liegt bei 90 % der Erkrankten vor dem 20. Lebensjahr. Therapeutisch ist Colchizin die First-line Therapie; bei Colchizin-Resistenz/Intoleranz sind IL-1 Inhibitoren empfohlen26, 27.
Mevalonatkinase-Defizienz (MKD)
Die MKD ist eine autosomal rezessiv vererbte Erkrankung, für die loss-of-function-Varianten im Mevalonatkinase (MVK)-Gen ursächlich sind28, 29. Bei der MKD handelt es sich um ein Krankheitsspektrum mit fliessenden Übergängen. Die Extreme des Spektrums repräsentieren das Hyper-IgD-Syndrom (HIDS), welches die milden Phänotypen zusammenfasst und die Mevalonazidurie (MA), welche die schweren Phänotypen bezeichnet. Der Schweregrad der MKD hängt mit der verbleibenden Aktivität der Mevalonatkinase zusammen30, 31. Beim HIDS ist meistens noch eine Restaktivität der MVK vorhanden, wohingegen bei der MA die Enzymaktivität fehlt oder unter der Nachweisgrenze liegt30, 31. Meist werden die Kinder bis Ende des 1. Lebensjahrs symptomatisch, Spätmanifestationen sind beschrieben32, 33. Charakteristisch für das HIDS sind 4–6 wöchentliche meist 3–7 Tage andauernde Krankheitsschübe32, 34. Die Schübe können in der frühen Kindheit hochfrequent sein (> 12 Schübe/Jahr) und nehmen mit steigendem Alter ab33. Typische Symptome sind Fieber (bis 40 °C), Schüttelfrost, schmerzhafte zervikale oder generalisierte Lymphadenopathie, gastrointestinale und muskoloskelettale Beschwerden sowie mukokutane Symptome (v. a. makulopapulöse Exantheme und Aphten)32–35. Bei der MA kommt es neben den beim HIDS bereits beschriebenen autoinflammatorischen Schüben zu dauerhaften Symptomen, zu denen eine schwere Entwicklungsverzögerung, eine psychomotorische Retardierung und eine Gedeihstörung wie auch faziale Dysmorphien, Hepatosplenomegalie, zerebelläre Ataxie, muskuläre Hypotonie, rezidivierende Konjunktividen, Katarakt und Uveitis zählen33, 36, 37.
Tumornekrosefaktor-Rezeptor-assoziiertes periodisches Syndrom (TRAPS)
Beim TRAPS sind autosomal-dominant vererbte Varianten oder de novo Varianten im TNFRSF1A-Gen, welches für den Tumornekrosefaktor-Rezeptor 1 (TNFR1) codieren, krankheitsursächlich2, 38, 39. Die Erstbeschreibung erfolgte unter dem Begriff «Familial Hibernian Fever»40. TRAPS manifestiert sich oft im Kleinkindalter, kann aber auch im Erwachsenenalter auftreten 41. Das Phänotypen-Spektrum ist breit; schwere Verläufe finden sich vor allem bei Varianten, die Disulfid-Brücken im TNFR1 betreffen42. Typisch sind schubartige, unregelmässige oft febrile Verläufe mit Dauer von 1–3 Wochen, allerdings sind auch kontinuierliche Krankheitsverläufe beschrieben41. Weitere charakteristische Krankheitssymptome sind wandernde Myalgien, Hautausschläge (bei Schubbeginn oft makulopapulös), Konjunktivitis, periorbitale Ödeme, Kopfschmerzen und Lymphadenopathie41, 43. Besonders im Kindesalter werden Bauchschmerzen und Lymphadenopathie berichtet41, 44. Zur Therapie des TRAPS werden als Mittel der Wahl IL-1 Inhibitoren verwendet, mitunter können Glukokortikoide im Schub zur Durchbrechung erwogen werden19, 45.
Haploinsuffizienz A20 (HA20)
Erstmalig wurde die Haploinsuffizienz A20 (HA20) im Jahr 2016 beschrieben46. Ursächlich für die HA20 sind autosomal-dominant vererbte loss-of-function Mutationen im TNFAIP3-Gen, welches für das A20 Protein kodiert und wodurch es zu einer unkontrollierten Aktivierung des NF-kB Weges mit vermehrter Produktion proinflammatorischer Zytokine kommt46. Charakteristisch sind frühe systemische Inflammation mit rezidivierenden oralen, genitalen und/oder gastrointestinalen Apthen/Ulzera47. Auf Grund der Ähnlichkeit zum M. Behçet sollte daher differentialdiagnostisch insbesondere bei jungen Patienten mit positiver Familienanamnese an die HA20 gedacht werden48. Neben den Behçet-ähnlichen Symptomen wie Exanthemen, Fieber, Augenentzündungen (z. B. anteriore Uveitis), und Polyarthritis/Arthralgien46, 47 sind variable entzündliche und autoimmune Präsentationen beobachtet worden, z. B. gastrointestinale Manifestationen, welche einer chronisch-entzündlichen-Darmerkrankung gleichen, oder Thyroiditiden sowie fluktuierende Autoantikörper (z. B. antinukleäre Antikörper, anti-dsDNA). Evidenzbasierte Therapieempfehlungen fehlen. Vielfach sind Zytokin-Inhibitoren (z. B. Tumornekrosefaktor (TNF)-Inhibitoren, IL-1/IL-6 Inhibitoren) zur Entzündungskontrolle notwendig47.
Defizienz der Adenosin-Desaminase 2 (DADA 2)
DADA2 ist durch autosomal rezessive loss-of function Mutationen im CECR1-Gen (neu ADA2-Gen) bedingt, welches für die Adenosin Desaminase 2 kodiert. Erstmalig beschrieben zwei Forschergruppen 2014 DADA249, 50. Die meisten Patienten präsentieren sich mit ersten Symptomen bereits in der Kindheit. Die Klinik ist sehr weitreichend und umfasst verschiedene Phänotypen (neurologisch, vaskulär, inflammatorisch, immunologisch, hämatologisch), die sich zum Teil überlappen können51. Typische klinische Symptome sind rezidivierendes Fieber, Livedo reticularis, kutane Vaskulitis, wiederkehrende Strokes, nicht-zirrhotische portale Hypertension, Hepatomegalie, muskuloskelettale Beschwerden, Panuveitis/Opticusneuritis und Immundefizienz50, 52–54. Insbesondere das frühe Auftreten von lakunären Strokes sollte an ein DADA2 denken lassen und neben anderen Differentialdiagnosen von kindlichen Schlaganfällen bedacht werden. Laborchemisch kann eine Panzytopenie neben erhöhten Entzündungszeichen imponieren. Die Diagnose kann durch eine verminderte Enzymaktivität von ADA2 aus dem Blut und/oder dem Nachweis von zwei Mutationen im ADA2-Gen erfolgen. Eine frühe Therapie mit TNF-Inhibitoren kann für gewisse Formen eine erhebliche Morbidität verhindern55.
(Autoinflammation) PLCγ2-assoziierte Antikörperdefizienz und Immundysregulation (PLAID, APLAID)
PLCγ2-assoziierte Antikörperdefizienz und Immundysregulation (PLAID) wird durch Deletionen im PLCγ2-Gen verursacht56. Typisch für PLAID sind Kälte-induzierte Urtikaria, Antikörper-Defizienz und Immundysregulation mit erhöhter Infektanfälligkeit, Neigung zu atopischen Erkrankungen und Autoimmunität56, 57. Die Autoinflammation PLCγ2-assoziierte Antikörperdefizienz und Immundysregulation (APLAID) wird ebenfalls durch Mutationen im PLCγ2-Gen verursacht58. Neben systemischer und organbezogener Entzündung findet sich eine Immundefizienz. Typische Symptome sind erythematöse/vesikulopustuläre Hautausschläge, gastrointestinale Beschwerden, Augenentzündungen, rezidivierende Infektionen, interstitielle Pneumonitis und Arthralgien/Myalgien57. Evidenzbasierte Therapieempfehlungen existieren nicht. Die Therapie richtet sich nach den führenden Symptomen; Einsatz von intravenöser Immunglobulin-Substitution, Antihistaminika, Zytokin-Inhibition, Colchizin und systemischen Steroiden mit (teils inhomogenen) partiellem bis gutem Ansprechen wurden beschrieben57.
Periodisches Fieber, aphthöse Stomatitis, Pharyngitis und zervikale Adenitis (PFAPA)
Das PFAPA-Syndrom ist charakteristischerweise durch das periodische Auftreten von Fieberschüben mit aphthöser Stomatitis, Pharyngitis und zervikaler Adenitis über 3–6 Tage gekennzeichnet59. Oft treten die Krankheitsschübe regelmässig alle 2–8 Wochen (meist 3–4 wöchentlich) auf. Der Leidensdruck bei den Patienten und ihren Familien ist oftmals hoch12. Mitunter findet sich eine positive Familienanamnese für wiederkehrendes Fieber oder Tonsillitiden60. Typischerweise tritt die Erkrankung im Kindesalter auf und hat eine gute Prognose (oft Spontanremission), aber auch Erwachsene können erkranken61, 62. In der medikamentösen Therapie des PFAPA-Syndroms haben vor allem Glukokortikoide einen diagnostisch-therapeutischen Stellenwert; bei Steroid-Non response, hochfrequenten Schüben oder Schubverkürzung unter Glukokortikoiden kann Colchizin wirksam sein63–65. Eine Tonsillektomie wird in einzelnen Fällen diskutiert, die wenigen vorhandenen Studien zur Tonsillektomie bei PFAPA-Syndrom erlauben keine generellen Empfehlungen.
Der Weg zur Diagnose
Für eine Diagnosestellung ist es entscheidend, Aspekte in der Anamnese und bei der klinischen Untersuchung, die auf eine AID hinweisen können, zu erkennen. Insgesamt empfiehlt sich ein stufenweises Vorgehen. Dabei sollten am Anfang die Erhebung der Patienten- und Familienanamnese inklusive Ethnizität, eine körperliche Untersuchung und eine erste orientierende Laboruntersuchung stehen. Der Verdacht auf eine AID ergibt sich meist aus einer charakteristischen Anamnese, klinischen und laborchemischen Untersuchungsbefunden.
Anamnese
Im Zuge der Eigenanamnese sollten Symptombeginn, Dauer und Periodizität der Symptome erfragt werden. Da insbesondere bei IL-1 vermittelten AID Trigger Krankheitsschübe auslösen können, macht es Sinn, nach diesen zu fragen. Neben Kälteexposition (z. B. bei CAPS), zählen Stress, körperliche Anstrengung, Infektionen oder Impfungen als mögliche Trigger. Es sollte nach einem zugrundeliegenden Fiebermuster und Begleitsymptomen gefragt werden. Die Abgabe eines Beschwerdekalenders zur prospektiven Erfassung von Krankheitsschüben kann hilfreich sein und ermöglicht insbesondere auch nach Diagnosestellung ein späteres Monitoring der Krankheitsaktivität (z. B. Autoinflammatory Disease Activity Index (AIDAI))73. Bei der Familienanamnese sollte neben der Ethnie und möglicher Konsanguinität nach spezifischen Beschwerden wie frühem Hörverlust oder Nierenerkrankungen/-transplantationen gefragt werden, da mitunter erkrankte Familienangehörige noch nicht diagnostiziert worden sind.
Charakteristische klinische AID Symptome
Bei vielen AID kommt es zu schubweise auftretendem Fieber; dies kann aber auch fehlen. Weiter zeigen die Patienten Symptome, die im Zusammenhang mit Entzündungen des zentralen Nervensystems, der serösen Häute, Haut- und Schleimhaut, der inneren Organe sowie dem muskuloskelettalen System stehen. Hierzu gehören beispielsweise folgende Symptome:
- Allgemein: Fatigue, Gedeihstörung
- ZNS: Kopfschmerzen, Stroke, Schwerhörigkeit, aseptische Meningitis, Entwicklungsretardierung
- Augen: Konjunktivitis, Episkleritis, Uveitis, Papillenödem
- Haut/Schleimhäute: Exanthem, Erythem, Urtikaria, neutrophile Dermatitis, Ulzera, Aphthen, Pannikulitis
- Muskuloskelettal: Arthritis/Arthralgien,
Myositis/Myalgie, Kontrakturen, Fasziitis
Abdomen: Bauchschmerzen, Diarrhoen, Nausea, (Hepato-)splenomegalie - Seröse Häute und Lymphknoten: Lymphadenopathie, Pleuritis, Perikarditis, Peritonitis
Da viele AID mit schubweiser Entzündung assoziiert sind, können die Symptome in der Konsultation fehlen. Hilfreich ist es daher, die Patienten zu bitten, Symptome während der Krankheitsschübe, z. B. Hautausschläge oder Augenentzündungen, fotographisch zu dokumentieren, damit eine bessere Einordnung vorgenommen werden kann. Neben schubweisen Verläufen gibt es aber auch kontinuierliche Krankheitsverläufe mit persistierender Entzündung.
Laborchemische Hilfsuntersuchungen
Bei vielen AID (z.B. IL-1 vermittelte AID, PFAPA, DADA2) sind die klinischen Symptome mit laborchemischer Entzündung gekoppelt (z. B. Neutrophilie, erhöhte BSG, erhöhtes CRP, erhöhtes SAA). Bei schubweiser Entzündung normalisiert sich diese 7–14 Tage nach Schubende. Daher sollte im Schub und ausserhalb eines Krankheitsschubs ein Blutbild, CRP und SAA ergänzt um Leber- und Nierenwerte sowie ein Urinstatus mit Protein/Kreatinin Quotient bestimmt werden. Sollten auch ausserhalb des Krankheitsschubs erhöhte Entzündungswerte vorliegen, muss an eine subklinische Inflammation mit erhöhtem Risiko für eine Organschädigung gedacht werden.
Weitere Schritte bei Verdacht auf AID
Erhärtet sich der Verdacht auf eine AID durch Anamnese, körperliche Untersuchung und Entzündungswerte und sind andere Differentialdiagnosen ausgeschlossen, so sollte eine Zuweisung an ein Zentrum mit einer pädiatrischen/internistischen Rheumatologie erfolgen, welches über Expertise in der Diagnostik und Therapie von AID verfügt. Diagnosekriterien können helfen, eine AID Diagnose zu stellen oder einen spezifischen Verdacht zu äussern25, 74. Darüber hinaus gibt es aus dem Jahr 2019 von Gattorno et al. evidenzbasierte Klassifikationskriterien für IL-1 vermittelte AID und PFAPA59. Diese werden oft in der Klinik zur Einordnung von Symptomen herangezogen, auch wenn sie primär für die Forschung entwickelt wurden.
Mit erhärtetem Verdacht sollten notwendige Zusatzuntersuchungen, die je nach AID variieren, veranlasst werden. Diese Zusatzdiagnostik kann beispielsweise aus einem Hochton-Audiogramm, Hautbiopsien, Echokardiographie, Röntgen-, CT- oder MRT-Untersuchungen, Lumbalpunktion und Liquordiagnostik, Interferonsignaturen, wie auch spezifischen zytologischen, histologischen und laborchemischen Untersuchungen bestehen. Auch kann bei entsprechendem Verdacht auf eine AID im Zentrum eine molekulargenetische Untersuchung eingeleitet werden. Bei Patienten, die Kriterien für eine AID erfüllen, aber bei denen Phänotyp und Genotyp nicht übereinstimmen, kann ein Whole Exome Sequencing (WES) oder Whole Genome Sequencing (WGS) erwogen werden75. Die Kenntnis der Genvariante kann helfen, die Diagnose zu sichern und ermöglicht das Abschätzen des weiteren Erkrankungsverlaufs und das assoziierte Risiko für Organschäden.
Management von Autoinflammatorischen Erkrankungen
Nach Diagnosestellung einer AID sollte zeitnah eine Therapie zur Kontrolle der systemischen Entzündung eingeleitet werden. Nur so können Organschäden, wie zum Beispiel eine sekundäre Amyloidose, ein/weitere Stroke/s oder Schwerhörigkeit, aber auch eine reduzierte Lebensqualität verhindert werden. Die Wahl der medikamentösen Therapie hängt dabei von der genauen Diagnose der AID und der Krankheitsaktivität ab.
Zur Behandlung von AID stehen verschiedene Medikamentengruppen zur Verfügung. Dabei muss je nach Krankheitsaktivität und Risikofaktoren zwischen einer Bedarfstherapie im Schub und einer Dauertherapie unterschieden werden. Im Schub haben beispielsweise Steroide, nichtsteroidale Antirheumatika (NSAR) oder kurzwirksame IL-1 Inhibitoren einen Stellenwert. Ist eine Dauertherapie indiziert, werden zum Beispiel Colchizin, mittel- und langwirksame IL-1 Inhibitoren, IL-6 Inhibitoren, TNF-Inhibitoren und/oder Januskinase (JAK)-Inhibitoren eingesetzt19, 26, 55, 76. Mitunter muss bei therapierefraktären Verläufen eine Stammzelltransplantation in Erwägung gezogen werden45, 51. Ziel der medikamentösen Therapie ist es, eine Remission oder geringstmögliche Krankheitsaktivität zu erreichen; oft sind Anpassungen der Therapie über den Krankheitsverlauf nach dem treat-to-target (T2T) Prinzip nötig. Zur Erfassung der Krankheitsaktivität haben sich Beschwerdetagebücher, wie der «Autoinflammatory Disease Activity Index» (Infobox «AIDAI»), bewährt73. Neben der medikamentösen Therapie kann es sein, dass Patienten mit AID zusätzliche Hilfsmittel (Hörgeräte, Orthesen) und Physiotherapie/Ergotherapie benötigen. Auch kann je nach Persönlichkeit der Erkrankten und dem Leidensdruck eine frühe psychotherapeutische Anbindung und ein Angebot für Familientherapie wichtig sein oder eine Einbindung in Selbsthilfegruppen bzw. Patienten-Netzwerke. Die Therapie von Patienten mit AID sollte daher idealerweise multidisziplinär erfolgen.
Infobox «AIDAI»
Ein für IL-1-vermittelte AID validiertes Instrument zur Erfassung der Krankheitsaktivität ist der Autoinflammatory Disease Activity Index (AIDAI)73. Betroffene geben das Vorhandensein (1 = Punkt) oder das Fehlen (0 = Punkte) von Krankheitsspezifischen Symptomen (Fieber ≥ 38 °C, reduzierter Allgemeinzustand, Bauchschmerzen, Übelkeit/Erbrechen, Durchfall, Kopfschmerzen, Brustschmerzen, schmerzhafte Lymphknotenschwellung, Gelenk-/Muskelschmerz, Gelenkschwellungen, Augenentzündung, Hautausschlag) und den Einsatz von NSAIDs an.
Bei einem monatlichen Summen-Score < 9 gilt die Erkrankung als inaktiv, bei einem Score ≥ 9 als aktiv.
Zusammenfassung
In den letzten Jahren hat sich das Wissen im Bereich der Autoinflammation stetig erweitert. Für viele AID sind mittlerweile Diagnose- und Klassifikationskriterien sowie Spezifika im Erwachsenen- und Kindesalter bekannt. Durch die Entwicklung von Krankheitsaktivitäts-Messinstrumenten, ersten verfügbaren Biomarkern zur Messung des Therapieansprechens und standardisiertem Monitoring sowie der Verfügbarkeit von wirksamen Therapien hat sich die Prognose vieler Patienten mit AID verbessert. Internationale Kollaborationen haben dazu entscheidend beigetragen und werden auch in Zukunft essentiell sein, um die Versorgung der Patienten in diesem dynamischen Feld der Rheumatologie weiter zu optimieren.
Referenzen
- Broderick, L., et al., The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol, 2015. 10: p. 395–424
- McDermott, M.F., et al., Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell, 1999. 97(1): p. 133–44
- McGonagle, D. and M.F. McDermott, A proposed classification of the immunological diseases. PLoS Med, 2006. 3(8): p. e297
- Georgin-Lavialle, S., et al., [Clinical overview of auto-inflammatory diseases]. Rev Med Interne, 2018. 39(4): p. 214–232
- Papa, R., et al., Syndrome of Undifferentiated Recurrent Fever (SURF): An Emerging Group of Autoinflammatory Recurrent Fevers. J Clin Med, 2021. 10(9)
- Labrousse, M., et al., Mosaicism in autoinflammatory diseases: Cryopyrin-associated periodic syndromes (CAPS) and beyond. A systematic review. Crit Rev Clin Lab Sci, 2018. 55(6): p. 432–442
- Toplak, N., et al., An international registry on autoinflammatory diseases: the Eurofever experience. Ann Rheum Dis, 2012. 71(7): p. 1177–82
- Hausmann, J.S., et al., The patient journey to diagnosis and treatment of autoinflammatory diseases. Orphanet J Rare Dis, 2018. 13(1): p. 156
- Makay, B., N. Emiroglu, and E. Unsal, Depression and anxiety in children and adolescents with familial Mediterranean fever. Clin Rheumatol, 2010. 29(4): p. 375–9
- Obici, L. and G. Merlini, Amyloidosis in autoinflammatory syndromes. Autoimmun Rev, 2012. 12(1): p. 14–7
- Kuemmerle-Deschner, J.B., et al., Hearing loss in Muckle-Wells syndrome. Arthritis Rheum, 2013. 65(3): p. 824–31
- Grimwood, C., et al., Health-related quality of life in children with PFAPA syndrome. Orphanet J Rare Dis, 2018. 13(1): p. 132
- Erbis, G., et al., Living with autoinflammatory diseases: identifying unmet needs of children, adolescents and adults. Pediatr Rheumatol Online J, 2018. 16(1): p. 81
- Ben-Chetrit, E., et al., Consensus proposal for taxonomy and definition of the autoinflammatory diseases (AIDs): a Delphi study. Ann Rheum Dis, 2018. 77(11): p. 1558–1565
- Aksentijevich, I., et al., The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum, 2007. 56(4): p. 1273–1285
- Cordero, M.D., E. Alcocer-Gomez, and B. Ryffel, Gain of function mutation and inflammasome driven diseases in human and mouse models. J Autoimmun, 2018. 91: p. 13–22
- Kümmerle-Deschner, J.B., Cryopyrin-assoziiertes periodisches Syndrom. Z Rheumatol, 2012. 71(3): p. 199–208
- Welzel, T. and J.B. Kuemmerle-Deschner, Diagnosis and management of the cryopyrin-associated periodic syndromes (CAPS): What do we know today? J Clin Med, 2021. 10(1)
- Romano, M., et al., The 2021 EULAR/American College of Rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist. Ann Rheum Dis, 2022. 81(7): p. 907–921
- Tufan, A. and H.J. Lachmann, Familial Mediterranean fever, from pathogenesis to treatment: a contemporary review. Turk J Med Sci, 2020. 50(SI-2): p. 1591–1610
- Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell, 1997. 90(4): p. 797–807.
- French, FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet, 1997. 17(1): p. 25–31
- Omenetti, A., et al., Increased NLRP3-dependent interleukin 1beta secretion in patients with familial Mediterranean fever: correlation with MEFV genotype. Ann Rheum Dis, 2014. 73(2): p. 462–9
- Kallinich, T., B. Orak, and H. Wittkowski, [Role of genetics in familial Mediterranean fever]. Z Rheumatol, 2017. 76(4): p. 303–312
- Yalcinkaya, F., et al., A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology (Oxford), 2009. 48(4): p. 395–8
- Ozen, S., et al., EULAR recommendations for the management of familial Mediterranean fever. Ann Rheum Dis, 2016. 75(4): p. 644–51
- Ozen, S., et al., Defining colchicine resistance/intolerance in patients with familial Mediterranean fever: a modified-Delphi consensus approach. Rheumatology (Oxford), 2021. 60(8): p. 3799–3808
- Drenth, J.P., et al., Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet, 1999. 22(2): p. 178–81
- Houten, S.M., et al., Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet, 1999. 22(2): p. 175–7
- Mandey, S.H., et al., Mutational spectrum and genotype-phenotype correlations in mevalonate kinase deficiency. Hum Mutat, 2006. 27(8): p. 796–802
- Cuisset, L., et al., Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur J Hum Genet, 2001. 9(4): p. 260–6
- Ter Haar, N.M., et al., The Phenotype and Genotype of Mevalonate Kinase Deficiency: A Series of 114 Cases From the Eurofever Registry. Arthritis Rheumatol, 2016. 68(11): p. 2795–2805
- van der Hilst, J.C.H., et al., Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore), 2008. 87(6): p. 301–310
- Tanaka, T., et al., National survey of Japanese patients with mevalonate kinase deficiency reveals distinctive genetic and clinical characteristics. Mod Rheumatol, 2019. 29(1): p. 181–187
- Bader-Meunier, B., et al., Mevalonate kinase deficiency: a survey of 50 patients. Pediatrics, 2011. 128(1): p. e152–9
- Haas, D. and G.F. Hoffmann, Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis, 2006. 1(1): p. 13
- Hoffmann, G.F., et al., Clinical and biochemical phenotype in 11 patients with mevalonic aciduria. Pediatrics, 1993. 91(5): p. 915–21
- McDermott, M.F., et al., Linkage of familial Hibernian fever to chromosome 12p13. Am J Hum Genet, 1998. 62(6): p. 1446–51
- Mulley, J., et al., Gene localization for an autosomal dominant familial periodic fever to 12p13. Am J Hum Genet, 1998. 62(4): p. 884–9
- Williamson, L.M., et al., Familial Hibernian fever. Q J Med, 1982. 51(204): p. 469–80
- Lachmann, H.J., et al., The phenotype of TNF receptor-associated autoinflammatory syndrome (TRAPS) at presentation: a series of 158 cases from the Eurofever/EUROTRAPS international registry. Ann Rheum Dis, 2014. 73(12): p. 2160–7
- Nezos, A., et al., Molecular and clinical spectrum of four pedigrees of TRAPS in Greece: results from a national referral center. Rheumatology (Oxford), 2020. 59(6): p. 1241–1246
- Welzel, T., Kuemmerle-Deschner, J.B. (2022). TRAPS bei Kindern und Jugendlichen. In: Wagner, N., Dannecker, G., Kallinich, T. (eds) Pädiatrische Rheumatologie. Springer Reference Medizin. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-60410-6_56
- Gaggiano, C., et al., Clinical features at onset and genetic characterization of pediatric and adult patients with TNF-α receptor-associated periodic syndrome (TRAPS): a series of 80 cases from the AIDA Network. Mediators Inflamm, 2020. 2020: p. 8562485
- Ter Haar, N.M., et al., Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis, 2015. 74(9): p. 1636–44
- Zhou, Q., et al., Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet, 2016. 48(1): p. 67–73
- Aeschlimann, F.A., et al., A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis, 2018. 77(5): p. 728–735
- Ohnishi, H., et al., A Japanese family case with juvenile onset Behcet’s disease caused by TNFAIP3 mutation. Allergol Int, 2017. 66(1): p. 146–148
- Navon Elkan, P., et al., Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med, 2014. 370(10): p. 921–31
- Zhou, Q., et al., Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med, 2014. 370(10): p. 911–20
- Ombrello, A., Segel, R. (2019). Deficiency of Adenosine Deaminase 2 (DADA2). In: Hashkes, P., Laxer, R., Simon, A. (eds) Textbook of Autoinflammation. Springer, Cham
- Nanthapisal, S., et al., Deficiency of Adenosine Deaminase Type 2: A Description of Phenotype and Genotype in Fifteen Cases. Arthritis Rheumatol, 2016. 68(9): p. 2314–22
- Pichard, D.C., et al., Early-onset stroke, polyarteritis nodosa (PAN), and livedo racemosa. J Am Acad Dermatol, 2016. 75(2): p. 449–53
- Schepp, J., et al., Screening of 181 Patients With Antibody Deficiency for Deficiency of Adenosine Deaminase 2 Sheds New Light on the Disease in Adulthood. Arthritis Rheumatol, 2017. 69(8): p. 1689–1700
- Ombrello, A.K., et al., Treatment Strategies for Deficiency of Adenosine Deaminase 2. N Engl J Med, 2019. 380(16): p. 1582–1584
- Ombrello, M.J., et al., Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med, 2012. 366(4): p. 330–8
- Welzel, T., et al., Variant in the PLCG2 Gene May Cause a Phenotypic Overlap of APLAID/PLAID: Case Series and Literature Review. J Clin Med, 2022. 11(15)
- Zhou, Q., et al., A hypermorphic missense mutation in PLCG2, encoding phospholipase Cgamma2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet, 2012. 91(4): p. 713–20
- Gattorno, M., et al., Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis, 2019. 78(8): p. 1025–1032
- Hofer, M., et al., International periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis syndrome cohort: description of distinct phenotypes in 301 patients. Rheumatology (Oxford), 2014. 53(6): p. 1125–9
- Wurster, V.M., et al., Long-term follow-up of children with periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis syndrome. J Pediatr, 2011. 159(6): p. 958–64
- Vitale, A., et al., Demographic, clinical and therapeutic findings in a monocentric cohort of adult patients with suspected PFAPA syndrome. Clin Exp Rheumatol, 2016. 34(6 Suppl 102): p. 77–81
- Ter Haar, N., et al., Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis, 2013. 72(5): p. 678–85
- Welzel, T., et al., Colchicine Effectiveness and Safety in Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Adenitis. Front Pediatr, 2021. 9: p. 759664
- Gunes, M., S. Cekic, and S.S. Kilic, Is colchicine more effective to prevent periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis episodes in Mediterranean fever gene variants? Pediatr Int, 2017. 59(6): p. 655–660
- Padeh, S., et al., Familial Mediterranean Fever in the first two years of life: a unique phenotype of disease in evolution. J Pediatr, 2010. 156(6): p. 985–989
- Padeh, S., et al., Familial Mediterranean fever in children presenting with attacks of fever alone. J Rheumatol, 2010. 37(4): p. 865–9
- Pelagatti, M.A., et al., Long-term clinical profile of children with the low-penetrance R92Q mutation of the TNFRSF1A gene. Arthritis Rheum, 2011. 63(4): p. 1141–50
- Ruiz-Ortiz, E., et al., Disease Phenotype and Outcome Depending on the Age at Disease Onset in Patients Carrying the R92Q Low-Penetrance Variant in TNFRSF1A Gene. Front Immunol, 2017. 8: p. 299
- Rama, M., et al., A decision tree for the genetic diagnosis of deficiency of adenosine deaminase 2 (DADA2): a French reference centres experience. Eur J Hum Genet, 2018. 26(7): p. 960–971
- Rigante, D., et al., A comprehensive comparison between pediatric and adult patients with periodic fever, aphthous stomatitis, pharyngitis, and cervical adenopathy (PFAPA) syndrome. Clin Rheumatol, 2017. 36(2): p. 463–468.
- Sicignano, L.L., et al., Children and Adults with PFAPA Syndrome: Similarities and Divergences in a Real-Life Clinical Setting. Adv Ther, 2021. 38(2): p. 1078–1093
- Piram, M., et al., Validation of the auto-inflammatory diseases activity index (AIDAI) for hereditary recurrent fever syndromes. Ann Rheum Dis, 2014. 73(12): p. 2168–73
- Kuemmerle-Deschner, J.B., et al., Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis, 2017. 76(6): p. 942–947
- Schnappauf, O. and I. Aksentijevich, Current and future advances in genetic testing in systemic autoinflammatory diseases. Rheumatology (Oxford), 2019. 58(Suppl 6): p. vi44–vi55
- Cetin Gedik, K., et al., The 2021 European Alliance of Associations for Rheumatology/American College of Rheumatology points to consider for diagnosis and management of autoinflammatory type I interferonopathies: CANDLE/PRAAS, SAVI and AGS. Ann Rheum Dis, 2022. 81(5): p. 601–613.


